インスリンとレプチンの働きを抑制している脱リン酸化酵素 – PTPRJの役割とは
インスリンとレプチン
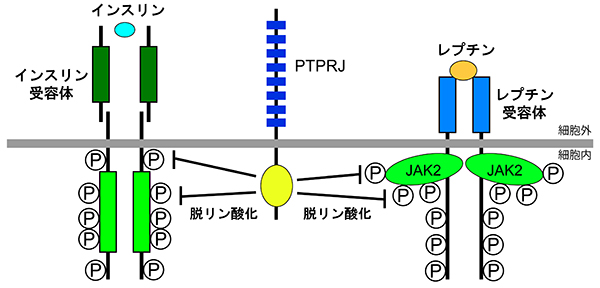
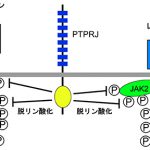
インスリンは、食後の血糖値の上昇に反応して膵島β細胞から血中に分泌されるホルモンで、血糖値を下げる働きをしています。インスリンが筋肉や脂肪細胞等の表面に存在するインスリン受容体に結合すると、細胞は血液中から細胞内への糖の取り込みを活性化し、エネルギー源として利用あるいは蓄積を始めます。インスリン受容体にインスリンが結合すると、その細胞内領域に存在する特定のチロシン残基を自己リン酸化することによって活性化し、続いて細胞内のシグナル分子をリン酸化することによって細胞内へ情報を伝えています。
一方、レプチンは脂肪細胞から分泌されるホルモンであり、脳内の弓状核という摂食行動をコントロールしている領域(摂食中枢)に作用して、摂食を強力に抑制します。レプチン受容体自身にチロシンリン酸化能はありませんが、細胞内領域にはJAK2というタンパク質リン酸化酵素(PTK)が会合しています。細胞表面に存在するレプチン受容体にレプチンが結合すると、JAK2は自身の特定のチロシン残基の自己リン酸化によって活性化し、次にレプチン受容体をリン酸化することによって、細胞内へレプチンの情報を伝えます。この情報によって、摂食中枢の神経細胞は活性化し摂食抑制を促します。
このように、インスリンやレプチンの受容体は機能的にはPTKであり、タンパク質のチロシンリン酸化を介した情報伝達を行っています。タンパク質のチロシンリン酸化の制御には、PTKだけでなく脱リン酸化酵素であるプロテインチロシンホスファターゼ(PTPs)が重要な役割を果たしています。PTPsは、膜タンパク質である受容体型(RPTPs)と細胞質に存在する非受容体型に分類されます。
PTPRJによるインスリンシグナルの制御
私たちは長年にわたりRPTPsの生理機能を明らかにする研究を進めてきました。最近、R3 RPTPサブファミリーに属するRPTPs(PTPRB, PTPRH, PTPRJ, およびPTPRO)がインスリン受容体を基質として、情報伝達に必要なリン酸化チロシンを脱リン酸化することによって、その活性化を抑制していることを見出しました。
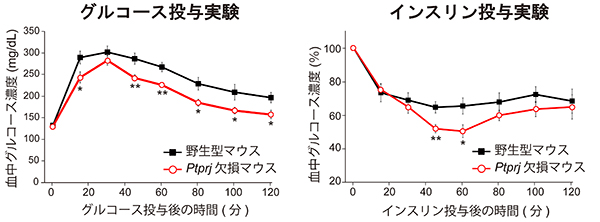
R3 RPTPsのひとつPTPRJは、インスリンの標的器官である、肝臓、筋肉、脂肪組織においてインスリン受容体と共に発現しています。そこでPTPRJの遺伝子欠損マウスを調べたところ、野生型マウスに比べて、インスリン受容体の活性が亢進していることが確認されました。マウスにグルコースを投与すると、Ptprj欠損マウスでは、野生型マウスに比べて、より速やかに血糖値が低下しました。一方、インスリンを投与すると、Ptprj欠損マウスでは血糖値の低下がより顕著でした。これらの結果は、Ptprj欠損マウスでは、PTPRJによるインスリン受容体に対する阻害が失われており、その結果、インスリンに対する感受性が亢進していることを示しています。
PTPRJによるレプチンシグナルの制御
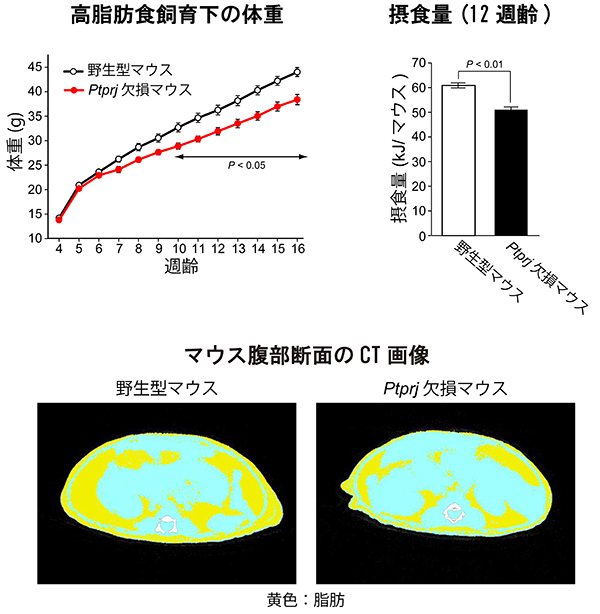
Ptprj欠損マウスは、野生型マウスと比べて、体長は変わりませんが、摂食量が少なく、低体重で、脂肪量が少ないことを見出しました。PTPRJは脳の摂食中枢の神経細胞においてレプチン受容体と共に発現しています。私たちは、PTPRJがレプチン受容体の活性を抑制しているのではないかと考え、そのメカニズムを解析しました。その結果、PTPRJが、レプチン受容体に会合したJAK2の自己リン酸化による活性化に重要なチロシン残基を脱リン酸化することによって、レプチンの働きを抑制していることが明らかになりました。Ptprj欠損マウスでは、PTPRJが無いために、レプチンによる摂食抑制の働きが亢進していると考えられます。そこで、マウス脳室にレプチンを投与したところ、野生型マウスに比べて、摂食量と体重が顕著に減少しました。
PTPRJのレプチン抵抗性への関与
肥満状態の人は、脂肪組織が多いためにレプチンのレベルは高い状態になっていますが、摂食は必ずしも抑制されていません。その理由は、レプチンが効きにくくなる、「レプチン抵抗性」と呼ばれる現象が起こるからです。レプチン抵抗性が生じるメカニズムはよくわかっておらず、その治療法も見つかっていません。
そこで、レプチン抵抗性の形成におけるPTPRJの関与について調べました。マウスを高脂肪食で2か月間飼育すると、レプチン抵抗性が形成されることが知られています。このとき、摂食中枢でPTPRJの発現が上昇していることがわかりました。
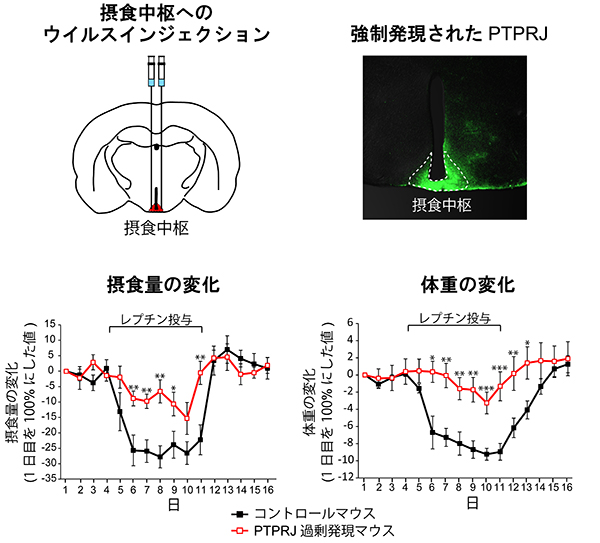
次に、高脂肪食で14週間飼育したマウスにレプチンを投与したところ、野生型マウスでは、レプチン抵抗性を発症しているために、摂食量と体重の減少はほとんど見られませんでした。一方、Ptprj欠損マウスでは、レプチン投与に応答して摂食量および体重の顕著な減少が見られ、レプチン抵抗性が生じていないことがわかりました。逆に、通常状態の野生型マウスの摂食中枢に、ウイルスベクターを用いて、人為的にPTPRJの発現を増加させると、レプチン抵抗性が誘導されました。このように、肥満にともない弓状核でPTPRJの発現が上昇することが、レプチン抵抗性形成の要因となっていることがわかりました。
最後に
PTPRJが、インスリン受容体とレプチン受容体の働きを抑制していることが明らかになったことにより、PTPRJを阻害する薬剤は、糖尿病(それに伴うインスリン抵抗性)とともに肥満(それに伴うレプチン抵抗性)を改善する治療薬となりうることがわかりました。
すなわち、PTPRJの活性を阻害することによって、少ないインスリンでもインスリン受容体が充分に活性化し、高血糖を改善することができると考えられます。また、同時に、レプチン受容体の活性化を促進し、肥満した人の食欲の亢進を改善できると考えられます。今後、PTPRJを標的とする薬剤が、糖尿病とともに肥満を改善する治療薬として開発されることが期待されます。
参考文献
Shintani T, Higashi S, Suzuki R, Takeuchi Y, Ikaga R, Yamazaki T, Kobayashi K and Noda M. (2017) PTPRJ inhibits leptin signaling, and induction of PTPRJ in the hypothalamus is a cause of the development of leptin resistance. Sci. Reports, 7, 11627. doi: 10.1038/s41598-017-12070-7.
Shintani T, Higashi S, Takeuchi Y, Gaudio E, Trapasso F, Fusco A and Noda M. (2015) The R3 receptor-like protein-tyrosine phosphatase subfamily inhibits insulin signaling by dephosphorylating the insulin receptor at specific sites. J. Biochem. 158, 235-243. doi: 10.1093/jb/mvv045.
この記事を書いた人
-
新谷隆史
基礎生物学研究所・統合神経生物学研究部門・准教授。1996年総合研究大学院大学生命科学科博士課程修了、博士(理学)。基礎生物学研究所・助手、助教を経て、2010年より現職。
どうして肥満するのか?肥満したら何がおきるのか?に興味を持って研究しています。
野田昌晴
基礎生物学研究所・統合神経生物学研究部門・教授。総合研究大学院大学・生命科学研究科・教授(併任)。1991年から現職。広く脳・神経系の様々な働きについて研究している。現在、日本プロテインホスファターゼ研究会の会長を務めている。
研究室ホームページ http://niwww3.nibb.ac.jp/
この投稿者の最近の記事
 研究成果2017年10月31日インスリンとレプチンの働きを抑制している脱リン酸化酵素 – PTPRJの役割とは
研究成果2017年10月31日インスリンとレプチンの働きを抑制している脱リン酸化酵素 – PTPRJの役割とは


