不良ミトコンドリアを分解する”マイトファジー” の制御因子を発見! – CRISPRによる順遺伝学スクリーニグで迫る
不良ミトコンドリアを分解する「マイトファジー」
細胞内のエネルギー産生をつかさどる細胞内小器官「ミトコンドリア」は、呼吸鎖と呼ばれるタンパク複合体によりアデノシン2リン酸(ADP)をリン酸化することで、エネルギー源であるアデノシン3リン酸(ATP)を産生します。このATPはアデニンヌクレオチド交換輸送体(ANT)によりミトコンドリアから細胞質へ運搬され、利用されます。また、ミトコンドリアは、酸化ストレスやカルシウム調節、細胞死の制御、糖・脂肪酸・アミノ酸の各種代謝にも関連しており、細胞の恒常性維持にとても重要であるといえます。
そのため、細胞は機能不全を起こした不良ミトコンドリアをオートファジー(自食作用)で分解する機能を有しています。この機能は、「マイトファジー」と呼ばれています。マイトファジーがうまく機能しないとミトコンドリアの機能も低下します。遺伝性パーキンソン病などの神経変性疾患では、マイトファジーの遺伝子異常が見られます。また、心不全や糖尿病をはじめとする疾患は、マイトファジー不全によるミトコンドリア機能の低下が関係していると考えられています。
マイトファジーの分子メカニズム
マイトファジーにはいくつかの機序があるのですが、そのなかで最もよく研究されているPINK1-Parkinによるマイトファジーの分子メカニズムを簡単に紹介します。PINK1とParkinは、ともに若年性パーキンソン病の原因遺伝子変異として知られていましたが、のちにマイトファジーの実行因子であることがわかりました。
まず、障害を受け膜電位が低下したミトコンドリアの外膜上にPINK1が蓄積します。そこにE3ユビキチンリガーゼであるParkinがリクルートされ、ミトコンドリア外膜タンパクがユビキチン化を受けます。次に、このユビキチンを足場としてユビキチン結合ドメインとオートファゴソームのLC3と結合するLIRを有するp62などのLC3レセプターが蓄積し、オートファゴソームを呼び込むことでミトコンドリアがオートファゴソームに取り込まれ分解されます。
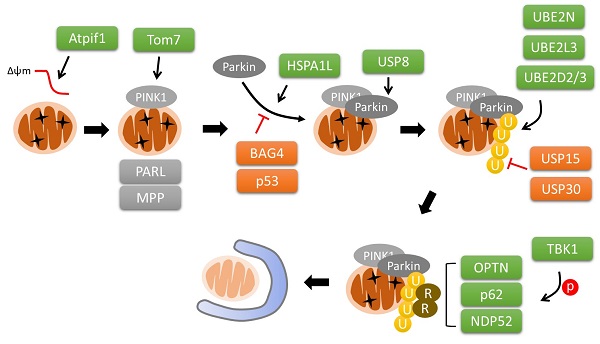
ミトコンドリアに障害が起きると膜電位が低下するが、ここにAtpif1が関与する。またPINK1は通常状態ではPARL、MPPの作用にて切断を受け、細胞質で分解されているが、膜電位の低下に反応して外膜に蓄積する際はTOM7が重要となる。Parkinがリクルートされる際はHSPA1Lが正に、p53、BAG4が負に制御し、ユビキチンペプチダーゼであるUSP8がParkinの活性化に関与する。Parkinがユビキチン化を行う際のE2リガーゼも同定されており、UBE2N、UBE2L3、UBE2D2、UBE2D3が関わる。またUSP15、USP30がユビキチン化を負に調節している。さらにp62タイプのLC3レセプターはTBK1によりリン酸化されることでLC3との結合力が強化される。
マイトファジーの関連遺伝子を同定 – CRISPRライブラリによる順遺伝学スクリーニグ
ジェノタイプ(遺伝子型)とフェノタイプ(表現型)の関連を見る研究において、フェノタイプからジェノタイプを調べる研究を順遺伝学研究といいます。反対に、ノックアウトマウスのフェノタイプを調べるような研究を逆遺伝学研究といいます。
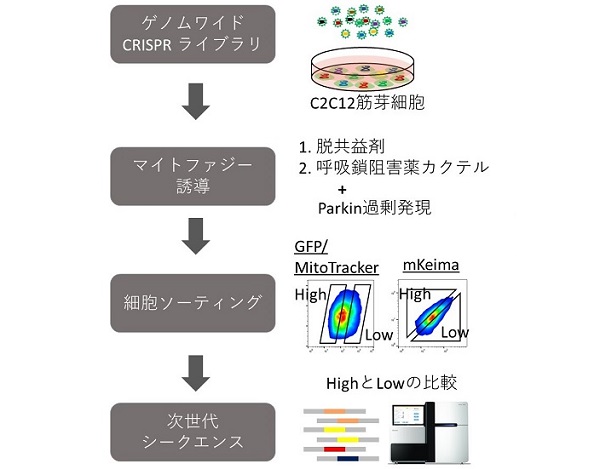
順遺伝学研究を行ううえで有力なツールのひとつとして、CRISPRライブラリがあります。これは、ひとつの遺伝子に対して4~10個のgRNAが設計され、合計8~20万個のgRNAがプールされたレンチウイルスベースのライブラリです。このウイルスライブラリをラージスケールで細胞に感染させることで、それぞれ異なる遺伝子がノックアウトされたヘテロな細胞集団を準備することができます。
今回の研究では、この細胞集団にマイトファジーを誘導し、マイトファジーが亢進している細胞集団(High)と低下している細胞集団(Low)をFACS(fluorescence activated cell sorting:蛍光活性化セルソーティング)で回収しました。そして、ゲノムDNAを抽出し、ディープシークエンスで集団に含まれるgRNAの差を解析することで、マイトファジーに関連する遺伝子を同定しました。
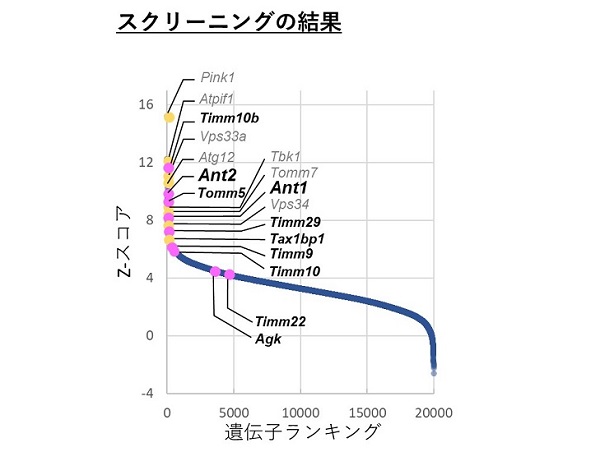
今回のスクリーニングでは、まず既知のマイトファジー、オートファジー関連遺伝子が数多くヒットし、スクリーニングが機能していることがわかりました。さらに、小胞の結合やエンドソームの成熟に必須であるHOPSやESCRTの構成因子が数多くヒットし、これらの重要性が再確認されました。
新規の因子としては、これまでにマイトファジーではLC3レセプターとしてOPTNとNDP52が必須と報告されていましたが、今回用いたマウスの筋芽細胞ではTax1BP1が必須でした。また、ミトコンドリアからATPを細胞質へ運搬する「ANT(アデニンヌクレオチド交換輸送体)」と、ミトコンドリア内膜にあるシャペロンタンパク「TIM22複合体」が、PINK1のミトコンドリア外膜への移行に必要であることがわかりました。
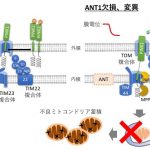
アデニンヌクレオチド交換輸送体(ANT)はTIM23を介してマイトファジーを制御する
ANTはミトコンドリアのATPと細胞質のADPを交換することでATPを細胞質へ運搬していますが、今回発見したANTのマイトファジー制御はこのはたらきとは独立したものでした。ANTはミトコンドリア内膜のタンパク輸送体であるTIM23複合体のTIM44と結合することでTIM23の輸送機能に関与し、マイトファジーを制御することがわかりました。
通常状態では、PINK1はTIM23に部分的に取り込まれ、PARLにより膜貫通ドメインが切断されることで細胞質へ放出され、プロテアゾームで分解されます。逆に、ミトコンドリア障害時はTIM23のゲートが閉鎖しPINK1がアクセスできなくなります。そのため膜貫通ドメインが保持され、PINK1はプロテアゾームで分解されずにTIM22のシャペロン機能の下で外膜に蓄積します。
ANTはこのTIM23のゲート閉鎖に重要で、ANTの欠損や一部の疾患遺伝子変異ではミトコンドリア障害時のTIM23のゲート閉鎖が不十分となります。そしてPINK1の膜貫通ドメインが切断され、プロテアゾームでの分解が生じるため、PINK1の蓄積が低下しマイトファジーの誘導が障害されることがわかりました。
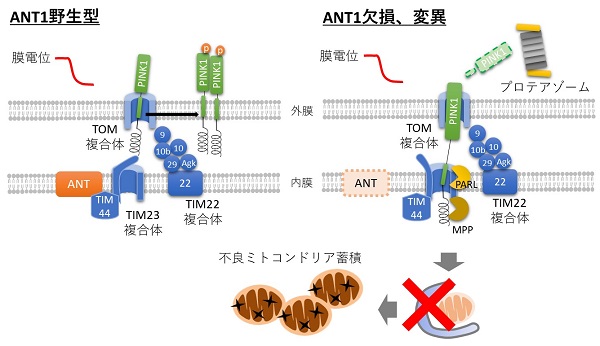
これまでに、ANT1の遺伝子異常によるミオパチーが報告されていましたが、一部の遺伝子異常ではADP/ATP交換輸送能は障害されておらず、機能的原因が不明でした。このような交換輸送能が正常な変異では、今回明らかとなったマイトファジーの誘導が障害されていることがわかり、今まで機能的原因が不明であったミオパチーはマイトファジー不全とそれによるミトコンドリア機能低下が原因となっていることが示唆されました。
今後の発展
ミトコンドリア機能の低下は神経変性疾患や心不全、糖尿病などの幅広い疾患と関連し、また老化にも関与していると考えられています。マイトファジーはミトコンドリア機能を正常に維持するために重要ですが、これらの疾患や老化においてマイトファジーが低下している報告が多くあります。そのためマイトファジーを亢進させることで病態の進行や、老化を抑えることができる可能性があります。これまでマイトファジーを活性化させる方法は確立されていないため、今後は本研究から明らかになった詳細なメカニズムも参考にしてマイトファジー活性化の新たな方法を開発していきたいと考えています。
参考文献
Hoshino A, Wang WJ, Wada S, McDermott-Roe C, Evans CS, Gosis B, Morley MP, Rathi KS, Li J, Li K, Yang S, McManus MJ, Bowman C, Potluri P, Levin M, Damrauer S, Wallace DC, Holzbaur ELF, Arany Z. “The ADP/ATP translocase drives mitophagy independent of nucleotide exchange.” Nature 575, 375-379 (2019)
doi: 10.1038/s41586-019-1667-4.
この記事を書いた人

- 2003年、京都府立医科大学医学部卒業。2011年、京都府立医科大学大学院医学研究科修了(2012年医学博士)。2014年よりペンシルバニア大学に留学し、2017年より京都府立医科大学循環器内科助教(現職)。大学院時代よりマイトファジーのメカニズムや病態生理的役割の解明に取り組んできました。現在はマイトファジー活性化の方法を模索すると共に、マイトファジーとミトコンドリア生合成の関係に関する研究、線維化に対する治療法の確立、またマイトファジーが関係する神経変性疾患にも対象を広げて研究に取り組んでいます。